生信宝典
生信宝典




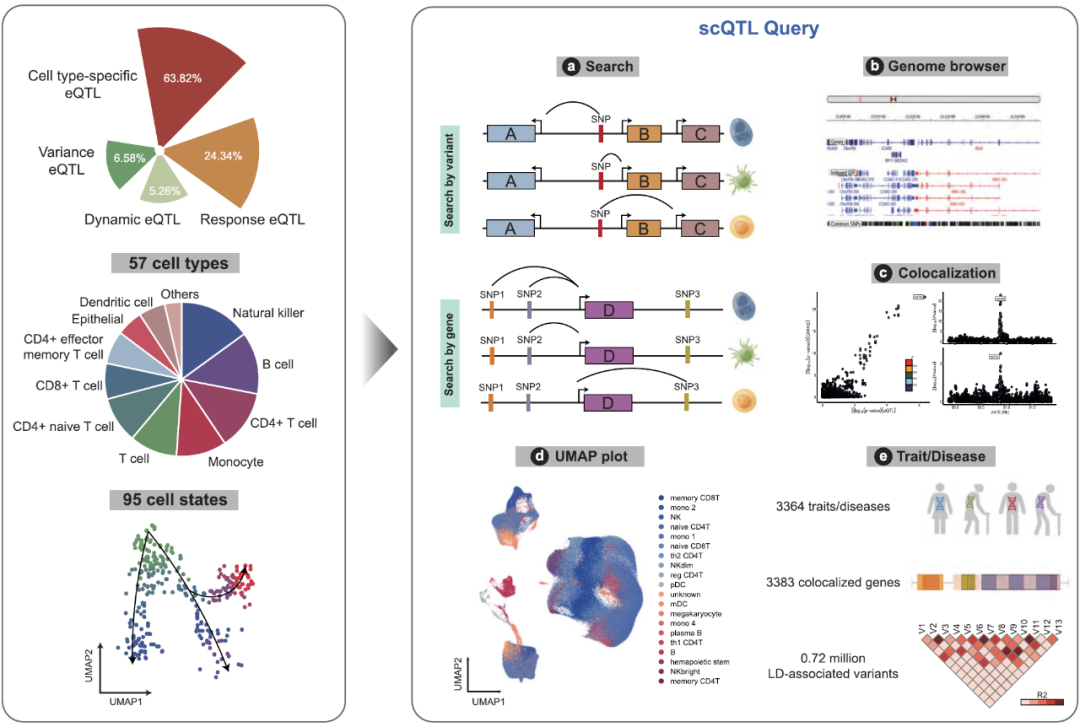
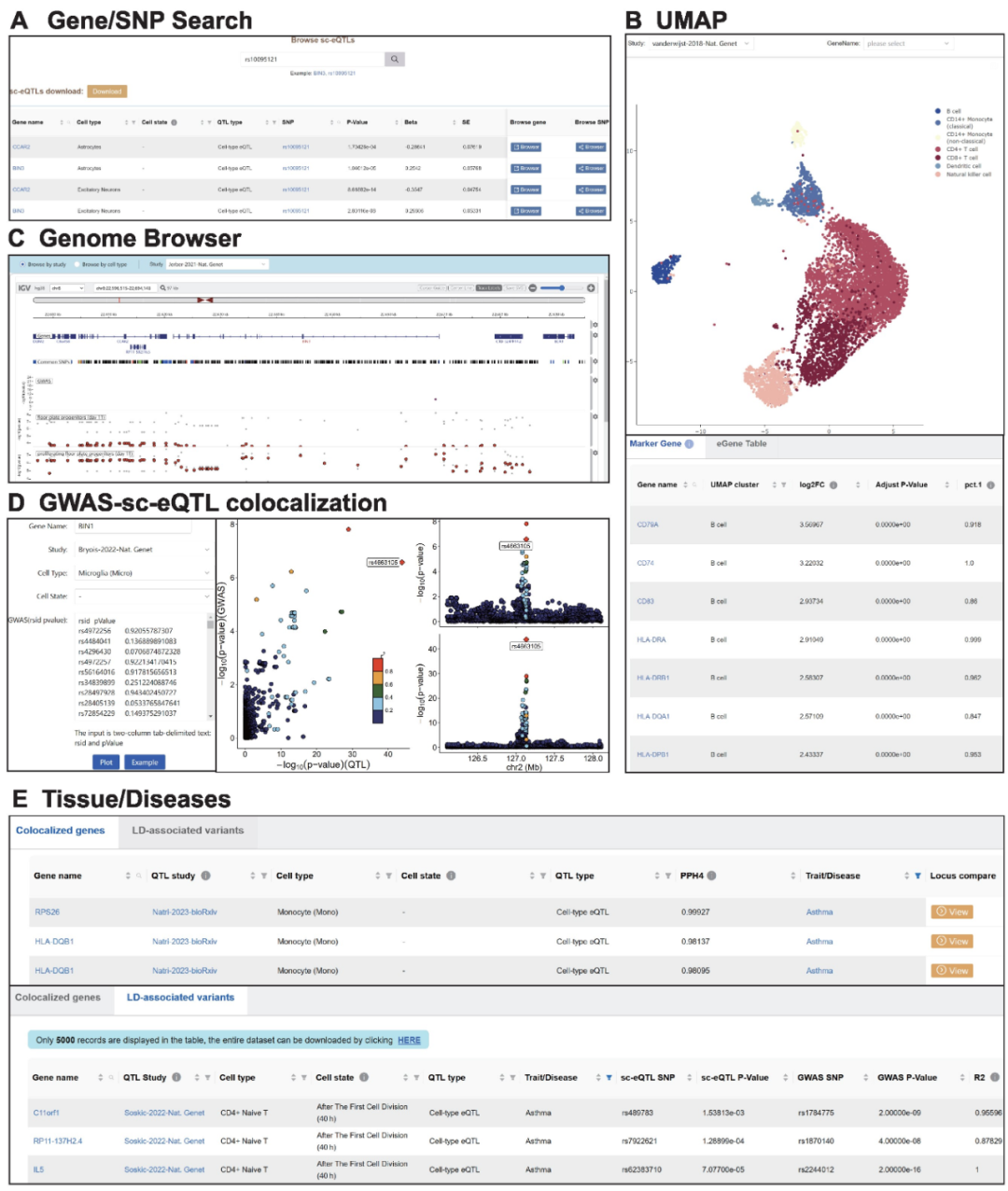
全基因组关联分析(Genome-Wide Association Studies,GWAS)已经确定了数千个与复杂性状和疾病相关的遗传位点,然而超过90%的GWAS风险位点位于非编码区域,这为确定疾病相关变异的分子机制带来了极大的挑战。研究表明,非编码区GWAS风险SNP(Single Nucleotide Polymorphisms)可以通过调控其上下游基因表达来影响疾病的进展[1]。因此,研究人员开发了SNP与基因表达变化关联的分析方法,即表达数量性状位点(expression Quantitative Trait Locus,eQTL)将疾病变异与靶基因联系起来。传统eQTL研究通常评估来自整个组织或样本的数百万个细胞的平均表达水平,掩盖了某些细胞类型或处于特定细胞状态的生物调节关系,只有20-50%的常见疾病关联基因被报道与eQTL共定位[2]。这意味着传统的eQTL在理解疾病相关变异方面有很大的局限性。近些年随着单细胞转录组测序技术的迅速发展,使得单细胞水平的eQTL(single-cell eQTL,sc-eQTL)分析成为可能,sc-eQTL可以在更高的分辨率下研究遗传变异对基因表达的调控关系[3,4]。整合大规模多来源的sc-eQTL数据集对于精细识别疾病因果变异和理解潜在的基因调控机制至关重要,然而,目前缺乏系统性研究多种复杂疾病在细胞水平的精细调控机制。
深圳湾实验室系统与物理生物学研究所李磊研究员为该论文通讯作者,课题组博士后丁若凡博士、硕士研究生王琪萱和研究助理龚力海为论文共同第一作者。数据库的构建得到了西班牙马德里生物工程、生物材料和纳米医学网络生物医学研究中心研究员Mireya Plass,宁波大学医学院廖奇教授的指导与帮助。
原文链接:
往期精品(点击图片直达文字对应教程)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|