 生信宝典
生信宝典





发表日期 :2022.9.23
摘要
背景
杨树物种具有不同的性别决定和频繁的性染色体转换。然而,与杨树相比,人们对柳树性别决定的多样性知之甚少,对杨树物种染色体更替的演化动力也了解甚少。在本文中,作者对S.chaenomeloides(钻天柳)和S.arbutifolia(腺柳)进行了性别鉴定,它们在7号染色体和15号染色体上分别有XY性别决定系统。
结果
基于性别决定区域的组装,作者发现柳树的性别决定机制可能与杨树有潜在的相似之处,都存在A 型细胞分裂素反应调节 (RR) 基因的完整或部分同源物。比较分析表明,在 柳属物种中至少发生了两次性染色体更替事件,一次保留了雄性异配的祖先模式,另一次将性别决定系统从 XY 变为 ZW,这可以解释为“deleterious mutation load”和“sexually antagonistic selection”理论模型。作者假设这些反复的更替使柳树物种的性染色体永远处于年轻状态,从而导致退化限制在一定程度。
结论
作者的研究结果进一步完善了杨柳科物种性染色体的演化轨迹,探索了驱动其性染色体反复更替的进化力量,为其他物种性染色体的研究提供了有价值的参考。
结果
基因组组装和注释
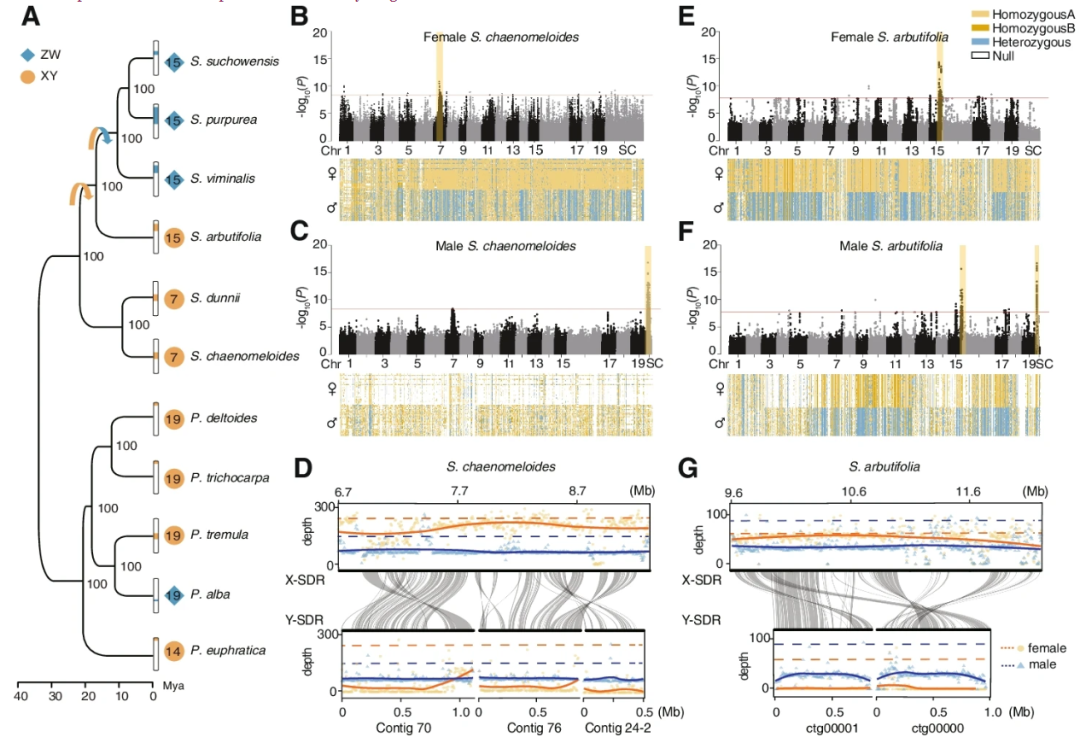
通过结合第三代长读测序、短读测序和染色体构象捕获(Hi-C)技术,作者对S.chaenomeloides和S.arbutifolia的雌性和雄性基因组进行了测序和组装。利用Illumina短reads在247× (98.7 Gb)覆盖范围内对一只雌性S.chaenomeloides的基因组进行了测序和组装,插入片段大小从350 bp到20 kb不等,并结合36× (14.9 Gb) PacBio测序数据进行了进一步的改进。将54.8 (169×) Gb的纳米孔长reads组装成contigs,然后使用Illumina短reads进行修正。两个基因组的染色体挂载率分别为90.59%和93.22%。对于S.chaenomeloides和S.arbutifolia的雄性基因组组装,生成57.9Gb (173×)和27.9 (94×) Gb的纳米孔长reads,并组装成N50大小分别为6.78M和2.65 Mb的contigs,其中98.61%和98.65%基于各自雌性参考基因组的共线关系锚定在19条染色体上。BUSCO分析显示,在1375个保守基因中,92.7 - 97.8%完全覆盖在4个基因组中。从Mercury获得的一致性质量值(QV)显示,QV在25.96 ~ 35.23之间,对应于装配一致性精度的99.75 ~ 99.96%。这些结果表明,基因组的组装具有高度的连续性、覆盖率和准确性。
基于转录本数据、同源性搜索和de novo预测,作者在S.chaenomeloides雄性基因组和S.arbutifolia雌性基因组中分别鉴定出27,595和29,609个蛋白质编码基因,分别包含95.5%和94.3%的完整保守基因。然后,作者在S.chaenomeloides、S.arbutifolia、其他9种已发表的杨柳科植物(包括4种柳树和5种杨树)和拟南芥中鉴定了1345个one to one的orthologs。利用这些同源基因进行系统发育分析,证实了柳属和杨属的单系关系(Fig. 1A)。与以往的研究一致,Protitea亚属的S.chaenomeloides和S.dunnii被鉴定为其余柳枝的姐妹,而Chosenia亚属的S.arbutifolia则是Vetrix亚属S.viminalis、S.purpurea和S.suchowensis`的姐妹。
由于s.anigra只有部分质体序列数据,作者根据3153 bp的质体序列对柳属进行了系统发育分析,结果表明s.anigra是s.achaenomeloides和s.adunnii的姐妹。分子年代测定表明,S.chaenomeloides和S.dunnii估计在大约22(Mya)前从其他柳树种类中分离出来,而下一个连续的分支S.arbutifolia估计在大约17 Mya产生(Fig. 1A)。
这一高分辨率的系统发育为后续研究其性别决定的动态进化历史提供了框架。
S.chaenomeloides 性别决定
对于S.chaenomeloides,作者对30个雄性个体和30个雌性个体进行了重测序,平均覆盖度为32×,以雌性个体的基因组为参考基因组,鉴定出了与性别显著相关的113个SNP(Fig. 1B)。大多数SNPs(92个,81.42%)在chr7的6.39 - 8.73 Mb之间。在这些SNP中,约74.2%的基因型在雄性中为杂合子,而在雌性中只有7.1%为杂合子。进一步分析表明,雌性的杂合子基因型几乎完全由3个个体引起,其中69.3%的基因型为杂合子,表明X和Y-SDR之间偶尔发生重组事件,这种现象在杨柳科其他物种中也有发现。当这些个体被排除在外时,在雌性只有0.2%是杂合子。因此,这个结果与XY系统中雄性染色体为异配性相一致。作者还发现其余与性别相关的SNP分散在染色体的不同位置(Fig. 1B)。这种模式也在其他杨柳科物种中存在,这是因为参考基因组来自一个同源性个体(XX或ZZ)和Y-(或W-)有限基因错位。因此,又将重测序数据重新比对到组装的雄性参考基因组上,发现了132个性别相关的SNP,所有这些SNP都位于三个未锚定的contigs上(Fig. 1C)。令人惊讶的是,作者发现在132个与性别相关的SNP中,73.36%的基因型在雌性中缺失,而66.29%的基因型在雄性中纯合。这一结果最有可能的原因是,这三个contigs是S.chaenomeloides Y染色体所独有的,因此没有来自雌性(XX)的reads映射到这些区域,这导致了只有来自表现出半合子Y基因型的雄性个体的Y特定区域的reads对齐。与这一解释相一致的是,作者发现这三个contigs与雌性基因组chr7上的性别相关区域高度相似。此外,这些contigs与雄性参考基因组的chr7的6.70 - 8.93 Mb具有广泛的共线性,该区域包含321个SNP,尽管经过多次测试和Bonferroni校正后,其性别相关p值小于1.0×10−7,并不显著。这些SNP在雌性中纯合子的占88.45%,在雄性中杂合子的占95.72%,符合XY性别决定。进一步支持作者的推论的是:所有这些区域在雄性中显示的测序深度是其他基因组区域的一半(Fig. 1D)。这些结果有力地表明,作者已经分别组装了X染色体和Y染色体的片段。根据性别相关SNPs的分离模式和测序深度分布,将雄性chr7对应的区域(约6.7 - 9.3 Mb)称为X-SDR,将三个跨度约2.5 Mb的离散contigs称为Y-SDR。
总的来说,结果清楚地表明,S.chaenomeloides具有一个XY性别决定系统,SDR位于chr7的非末端区域。
S.arbutifolia 性别决定
对于s.arbutifolia,作者对26个雄性个体和30个雌性个体的基因组进行了重测序,平均覆盖率为38×,并采用了类似的GWAS策略来识别性别相关的SNPs。以S.arbutifolia雄性基因组为参照,共鉴定出126个与性别显著相关的SNPs (Bonferroni校正后α < 0.05)(Fig. 1F)。其中61个(48.41%)发生在chr15上,53个(42.06%)发生在与S.purpurea基因组chr15共线的一个未锚定contig上。在这些SNP中,约89.84%的基因型在雌性中为纯合子,70.45%在雄性中为杂合子,这一模式与XY性别决定系统基本一致。参照等位基因在雄性中纯合的比例为20.63%。与上述结果相似,这一观察结果可能是由于雄性参照体中X-Y嵌合组合的存在。当以雌性基因组为参考时,XY性别确定系统的SDR位于chr15上也得到了确认 (Fig. 1E)。由于在雄性基因组中没有发现明显的Y特异性序列,作者将雄性特异性k-mer与WhatHap进行的基于read的组装结合,将Y-SDR重建为两个总长度约1.8 Mb的contigs。
这个结果得到了雄性测序深度和与雌性性别相关区域共线性的支持 (Fig. 1G)。
比较两个柳树物种中的 Y‑SDRs

为了深入了解柳树特异编码基因的组成和进化史,作者在S.chaenomeloides和S.arbutifolia分别预测到了 Y-SDR中的78个和91个蛋白编码基因。通过与相应的X-SDRs对比,作者发现这些基因所在的区域也富含大量串联重复基因、常染色体易位基因和SDR特异性基因。然而,大多数Y特异基因的功能不明,且两种柳树Y-SDR之间没有同源的蛋白编码基因。接下来,作者估计了共享的X-Yhomologs之间的同义替换(Ks),以检验这两个物种是否存在差异程度不同的evolutionary strata。S.chaenomeloides共鉴定出37对X-Yhomologs,Ks中位数为0.033±0.014 SE(Fig. 2A)。52对S.arbutifoliaX-Yhomologs间的Ks值为0.020±0.033 SE (Fig. 2A)。没有发现明显的证据支持这两个物种中strata存在。然而,S.arbutifolia的X-Yhomologs之间的差异较小,表明其SDR比S.chaenomeloides进化得更晚。此外,基于同源性搜索和严格的相似度和覆盖率过滤标准,识别了位于柳树的SDRs上的假基因。作者发现,S.chaenomeloides和S.arbutifolia Y-SDR上的假基因数量(分别为22和14)大于X-SDR上的假基因数量(分别为8和8)。与X-Yhomologs相比,这些假基因表现出较高的非同义替换比(Ka/Ks),表明它们经历了宽松的选择压力或中性进化。有趣的是,S.chaenomeloides假基因的Ks分布显著高于X-Yhomologs,略高于S.chaenomeloides与S.arbutifolia的差异 (Fig. 2A)。同样,S.arbutifolia假基因的Ks值略高于S.arbutifolia和S.purpurea的 差异 (Fig. 2A)。
这些观察结果表明,假基因中核苷酸取代率的增加,并且揭示了SDR在起源后逐渐和持续的分化。
Y‑SDRs 遗传退化
SDR的重组抑制具有重要的进化作用,除了丧失基因活性外,还包括容纳重复元素和有害突变的积累。为了更详细地研究退化,作者首先在这些SDR区域注释了重复序列。结果表明,S.chaenomeloides和S.arbutifolia中X-和Y-SDR的重复序列含量均高于伪常染色体区域(PARs)和其他常染色体区域,特别是长末端重复序列(LTR)逆转录转座子的含量较高 (Fig. 2B)。进一步的分析发现,在过去的3000万年和2300万年中,S.chaenomeloides和S.arbutifolia的Y-SDR中,分别有71和60个完整的LTRs(Fig. 2C)。这些结果表明,在SDRs建立后,转座元件在这些区域迅速积累。同时,为了探索Y-SDRs上mutation load的积累,作者使用PolyPhen2和PROVEAN,(基于不同植物谱系对该位点的保护),将编码区域的变异碱基分为有害(DEL)、容忍(可能是轻微有害,TOL)或同义(SYN,假设是中性选择)。以SYN变异选为中性选择的参照,S.chaenomeloidesY-SDR中DEL和TOL变异的比例显著高于X-SDR和PAR(Fig. 2D)。但在S.arbutifolia中未发现这种模式,其与X-SDR和PAR基因相比,Y-SDR包含更多的TOL变异,但不包含更多的DEL变异(Fig. 2D)。
这表明S.chaenomeloides的Y-SDR比S.arbutifolia经历了更多的遗传退化。
柳树中的性别决定机制
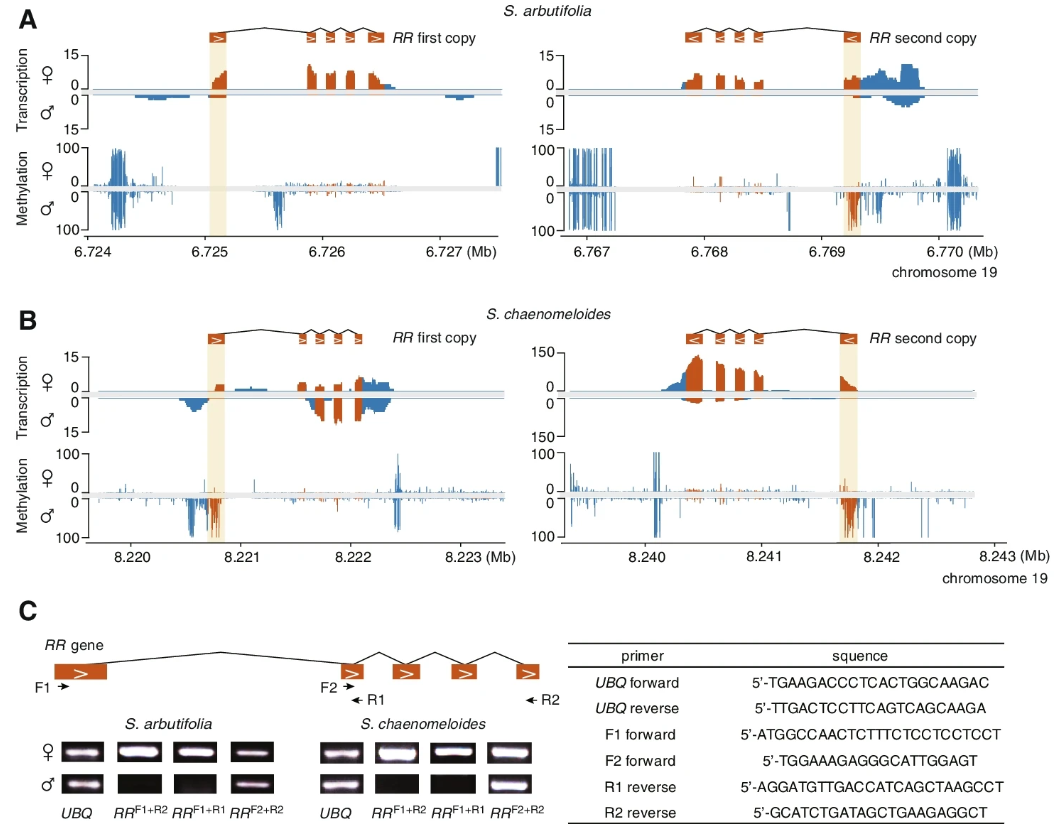
近年来的研究表明,RR基因是杨树性别决定的主要调控因子,表达时可驱动雌性发育,抑制时可驱动雄性发育。为了研究S.chaenomeloides和S.arbutifolia是否存在相同的性别决定机制,作者研究了RR基因在其雌雄花蕾中的完整表达模式。
两个物种的chr19(常染色体)上发现了两个完整的RR基因,核苷酸序列相似性超过98.6%。RNA-seq数据证实了两种RR基因在S.arbutifolia花蕾中都有雌性特异性表达,这与RR基因沉默对雄花发育很重要的假设是一致的 (Fig. 3A)。相比之下,RR基因的两个拷贝,外显子均在S.chaenomeloides雄花花蕾中表达,但在第二个拷贝中表达量明显低于雌花 (Fig. 3B)。作者验证了RNA-seq reads的对齐,发现两个RR基因的第一个外显子在雄性中被特异性沉默,这一结果被RTPCR证实 (Fig. 3C)。此外,作者还发现了第一外显子和上游区域,S.chaenomeloides雄性花蕾甲基化水平显著升高 (Fig. 3A, B)。这表明RR基因的雄性特异性沉默可能是由于选择性剪接或转录起始位点的转移,这与相应区域的高DNA甲基化有关。为了验证在杨树中发现的RR部分副本作为雌性抑制子抑制完整RR基因的机制是否也存在于XY柳树中,作者搜索了S.chaenomeloides和S.arbutifolia Y-SDR上RR基因的同源序列。不出所料,在两个物种中都发现了RR基因的部分副本 (Fig. 4A)。在两个物种中共发现了9个重复基因,其中7个与完整的S.chaenomeloides RR基因的第1外显子同源,2个与第4外显子同源。在S.arbutifolia中,另一个副本与第4和第5外显子同源。与雄花花蕾中缺乏完整RR基因的表达一致(Fig. 3A–C)。
小RNA-seq检测到与这些部分重复基因同源的小RNA,可能抑制完整RR基因的表达(Fig. 4A)。重要的是,在两个物种的RR部分副本周围发现了两个完整的LTRs (Fig. 2C),这表明它们可能参与了RR片段向SDY的转位。在S.chaenomeloides中,这两个LTRs的插入时间分别为21Mya和4 Mya,插入时间较早,发生于其与杨树的分化之后,但接近于杨树的分化。而在S.arbutifolia中,插入时间估计分别为13和9 Mya,均发生在S.arbutifolia与S.chaenomeloides分离之后,进一步证实S.arbutifolia的SDR起源较晚S.chaenomeloides(Fig. 2C)。
应该指出的是,由于大部分RR部分副本周围缺乏完整的LTR和Y-SDR的持续退化,这些估计时间可能被低估了。总的来说,这些观察结果一致支持柳树和杨树物种性别决定机制的相似性,都涉及完整和部分的RR序列。
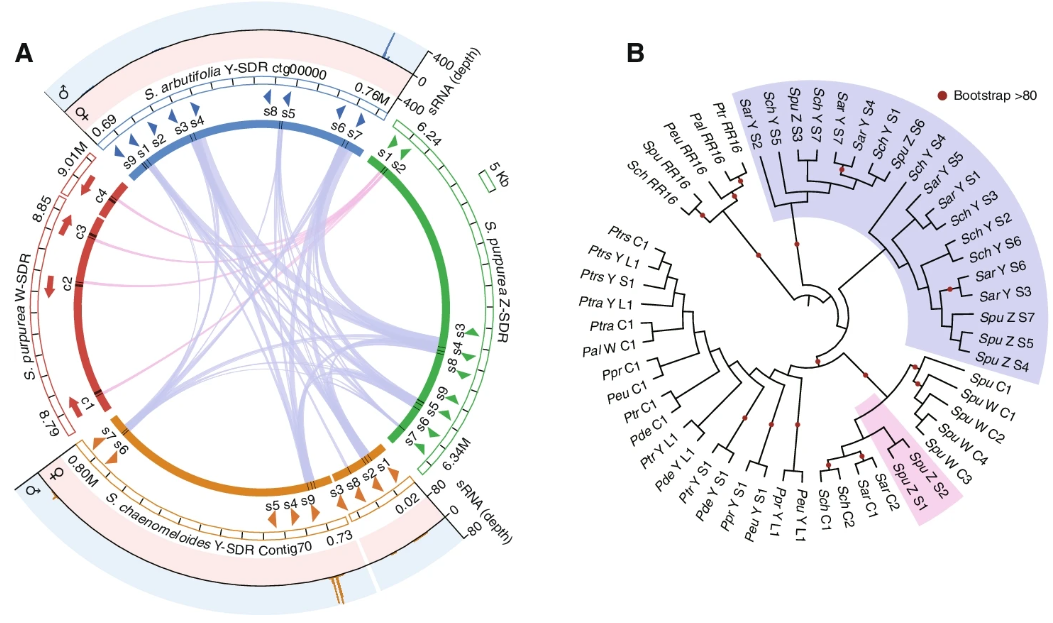
令人惊讶的是,作者还在S.purpurea的Z-linked区域发现了9个RR部分副本,其中两个之前已经发现,并与位于S.purrea W-SDR上的4个RR完整基因副本具有广泛的同源性,而其他7个副本与第一个外显子同源。“SpuZ:S8”到第4外显子和“SpuZ:S9”到第4和第5外显子与S.chaenomeloides和S.arbutifolia RR部分副本的相似性高于“SpuZ:S1-S2”(Fig. 4A)。通过对已知的杨科植物RR完整和部分拷贝的系统发育关系分析,发现分别来自S.chaenomeloides、S.arbutifolia和S.purpurea的部分拷贝“SchY:S1-S7”、“Sar:S1-S7”和“SpuZ:S3-S7”聚在一起,系统发育过程表明它们的起源早于柳树和杨树RR完整基因的分化(Fig. 4B)。另外,三种柳树的RR基因中均存在SpuZ:S1和SpuZ:S2基因,推测其独立来源较近。
综上所述,这些结果表明,在杨柳物种的多样性过程中,产生小RNA的Y-SDR在较早的进化过程中产生,而柳属物种的性染色体翻转可能与杨属物种的RR序列易位相同。
讨论
在本文中,作者确定了两种柳树的性别决定,并证实与杨树一样,柳树也表现出快速的rate of sex chromosome turnovers。虽然之前的研究已经报道了柳属物种中 chr7上的XY 系统和chr15上的ZW系统,但本文报告 chr15 上的新XY系统并在 XY物种中组装 了Y-SDR,可以更深入的研究柳属物种性染色体的进化历史。与先前的研究一致,两种柳树的Y-SDR均大于杨树。这一观察结果可以解释为柳树SDR与着丝粒的部分重叠,在控制性别决定的基因和影响柳树中更明显的性二态性的基因之间选择重组抑制,或数量的差异。影响昆虫授粉柳树和风授粉杨树之间授粉模式相关性二态性的基因。无论这些推测的结果如何,本文都揭示了柳树物种性别决定的丰富多样性,并为进一步研究系统发育框架下性染色体的动态变化奠定了基础。研究结果还表明,杨树和柳树可能具有相似的性别决定机制,包括完整的RR 基因和部分RR重复。这个猜测得到了两个柳树种的 Y-SDR 上存在 RR 部分重复和 RR 基因的雌性特异性表达的支持。(Figs. 3 and 4)
此外,这些 RR 部分重复的系统发育位置表明它们出现在 Salix 和 Populus 属的分歧之前。(由于序列短,这种模式的 bootstrap support很低)(Fig. 4B)。尽管如此,这种机制在柳树中仍然存在争议,因为 RR 基因和部分重复在S.purpurea的 W 和 Z 染色体上共存。未来对其他柳树物种的研究或详细的功能研究应该为这种机制的普遍适用性提供重要的见解。性别决定的比较表明,柳树品种多样化过程中至少发生了两次更替事件(Figs. 1A and 5)。物种系统发育关系、基因的分歧和 SDR 中 LTR 插入的时间,以及 RR 部分重复的古老起源, (Figs. 1A, 2A, C, and 4B)支持 chr7 上XY SDR作为柳属的祖先(推测)。第一次性染色体更新发生在chr7 (S. chaenomeloides) 到 chr15 (S. arbutifolia),延续了XY系统。第二次性染色体更新事件发生在 chr15 上的异配从 XY (S.arbutifolia) 变为 ZW (S.purpurea) (Figs. 1A and 5)。性染色体的转变本质上是新的或旧的性别决定位点的移动,研究结果表明Salix 的性染色体更替可能与 RR 序列有关。RR 部分重复在第一次转换事件中从 chr7 易位到 chr15,第二次转换事件涉及常染色体 RR 完整基因从 chr19 到 chr15 的易位 (Fig. 5)。在其他一些物种中也提出了类似的主要性别决定基因易位到常染色体导致新的性染色体出现。一些理论已经预测了推动性染色体转变的进化力量,本文的研究揭示了哪些因素可能推动了 Salix中性染色的转变。deleterious mutation模型假设有害突变在非重组区域的积累会降低异配性别的适应度,从而促进性染色体转变以避免genetic load。观察发现,S.chaenomeloides Y-SDR 携带比 S.arbutifolia 更多的假基因和有害突变(Fig. 2D),这表明S.chaenomeloides的遗传退化更为先进Y-SDR 可能比稍年轻的 S.arbutifolia Y-SDR 更早开始。这种模式与导致 chr15 Y-SDR 部分受到青睐的想法是一致的,因为这一举措从 SDR 中清除了有害基因座。因为 S.arbutifolia 和 S.chaenomeloides 的性染色体都是 XY 系统,作者推测这是ancestral state并且 ZW-SDR 系统也来自 XY。以前的大多数工作都认为这是不同异配性别系统最常见的过渡方向。例如,在一些鱼类和两栖动物中的实验证实,W 在上位性上优于 Y ,并且在多个物种中也观察到了从 XY 到 ZW 的性转变。更有趣的是,S.arbutifolia的 Y-SDR 和S.purpurea的 Z-SDR 中高度相似的 RR 部分重复表明 Y 在第二次转变中变成了 Z(Figs. 4A and 5)。以这种方式衍生的 Z-SDR 在纯合时必须是可行的,这可能是在第一次转换后新的 chr15 Y 出现后不久的情况。或者,如果 chr15 Y-SDR 持续存在并积累genetic load,则在 W 的过渡和随后的进化过程中克服了这种负荷。模型表明,sexually antagonistic selection可以驱动异配子中的两种转换,而 SDR 保持在同一位置以及 SDR 基因组位置的变化。对于驱动 Salix的两个更新事件的sex-antagonistic selection,在 chr15 上进行sex-antagonistic selection的基因座必须与性别决定基因座(RR 序列)相关联,导致雄性比携带原始 Y 染色体的雄性具有更高的适应度。这一次,没有推动这一周转的性选择基因座的候选者。尽管deleterious mutations和sexually antagonistic selection模型可能导致Salix的性染色体更替,但其他进化力量,如genetic drift和biased sex ratios仍然无法排除出去。一些机制涉及特殊的生物学条件,例如小种群规模,可能需要进一步了解种群历史。同时,还应考虑多种机制的综合作用。
总的来说,作者的研究结果表明,杨柳科中性染色体的快速更新率是由多种进化力量驱动的,以保持其性染色体永远年轻。进一步探索驱动性染色体周转的机制需要更多物种的信息。
总结
本文分别在 S.chaenomeloides 和 S.arbutifolia 的 7 号和 15 号染色体上确定了一个 XY 性别决定系统,并发现它们可能与杨树具有相似的性别决定机制,涉及完整的 RR 基因和部分 RR 重复。进一步结合系统发育分析表明,柳树物种多样化过程中至少发生了两次更替事件,7XY到15XY和15XY到15ZW,这可以部分解释为“deleterious mutation load””和“sexually antagonistic selection”理论模型。这些结果进一步完善了杨柳科性别决定和性染色体进化的研究,表明重复的更替可能是柳树性染色体有限退化的原因。
课题组
课题组
四川大学生命科学学院 - 马涛

Reference
文章链接:https://genomebiology.biomedcentral.com/articles/10.1186/s13059-022-02769-w
往期精品(点击图片直达文字对应教程)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|




























后台回复“生信宝典福利第一波”或点击阅读原文获取教程合集




