GATK教程 / 体细胞短变异检测 (SNV+InDel)流程概览
共 5052字,需浏览 11分钟
· 2022-06-27
体细胞短变体检测 (SNV + InDel)
Somatic short variant discovery (SNVs + Indels)
目的
参考实现
另见文件:
gatk-master.zip
(How to) Call somatic mutations using GATK4 Mutect2.docx
链接:https://pan.baidu.com/s/1BTmmCM-mJ-hEA1wWNYDs4g
提取码:ysx4
预期的输入
该工作流程需要为每个输入的肿瘤和正常样本(Each input tumor and normal sample)提供BAM文件。输入Bam应按照《GATK数据预处理最佳实践 (GATK Best Practices for data pre-processing)》中的描述进行预处理:

https://gatk.broadinstitute.org/hc/en-us/articles/360035535912
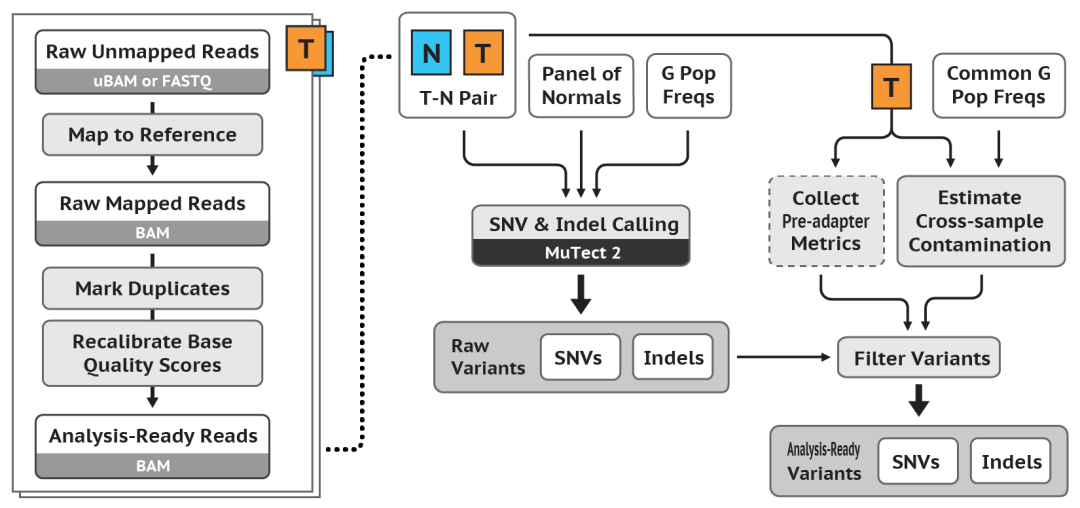
这个工作流有两个主要步骤:① 首先,生成大量的候选体细胞变异;② 然后,对它们进行筛选,以获得更有信心的体细胞变异调用集合 (A more confident set of somatic variant calls)。
检测候选变异(Call candidate variants)
工具:Mutect2
https://gatk.broadinstitute.org/hc/en-us/articles/5358824293659--Tool-Documentation-Index#Mutect2
计算污染
涉及的工具:GetPileupSummaries, CalculateContamination
https://gatk.broadinstitute.org/hc/en-us/articles/5358824293659--Tool-Documentation-Index#GetPileupSummaries
https://gatk.broadinstitute.org/hc/en-us/articles/5358824293659--Tool-Documentation-Index#CalculateContamination
Learn Orientation Bias Artifacts
工具:LearnReadOrientationModel
过滤变异 (Filter Variants)
工具包括:FilterMutectCalls
https://gatk.broadinstitute.org/hc/en-us/articles/5358824293659--Tool-Documentation-Index#FilterMutectCalls
FilterMutectCalls解释了相关的错误,也就是说,一个位点上所有的变异Reads都是由于某种共同的错误来源 (Due to some common source of error)的可能性。它通过几个硬过滤 (Several hard filters)来实现这一目标,以检测:“Alignment artifacts and probabilistic models for strand and orientation bias artifacts”(比如PCR产生的错误,会被后续的PCR循环传递下去,就出现“某种共同的错误来源”)、聚合酶滑移(Slippage artifacts,)、胚系变异和污染(都会出现“某种共同的错误来源”)。原因是:肿瘤体细胞突变相对随机,且通过超声法破碎后Reads长度大小不一。也因为如此,克隆性造血过程中自身白细胞等细胞中携带的突变,以及其它与肿瘤无关的体细胞突变将无法被过滤(除非做了患者自身配对的WBC实验设计及测序),因为这类突变也是:“相对随机,且通过超声法破碎后Reads长度大小不一”。
此外,FilterMutectCalls学习了一个关于肿瘤的整体SNV和InDel突变率和等位基因分数谱的贝叶斯模型,以改进Mutect2发出的对数几率 (A Bayesian model for the overall SNV and indel mutation rate and allele fraction spectrum of the tumor to refine the log odds emitted by Mutect2)。然后,它会自动设置一个过滤阈值,以优化:F评分、灵敏度和精度的调和平均值(F score, the harmonic mean of sensitivity and precision)。
注释变体
工具包括:Funcotator
https://gatk.broadinstitute.org/hc/en-us/articles/5358824293659--Tool-Documentation-Index#Funcotator
Funcotator生成一个“变异调用格式(Variant Call Format, VCF)”文件(在INFO字段中增加注释结果)或一个“变异注释格式(Mutation Annotation Format, MAF)”文件。
一些问题反馈
上面文字里的GetPileupSummaries和CalculateContamination怎么用还不清楚。应该先使用哪个工具?或者它们应该同时使用吗?
往期精品(点击图片直达文字对应教程)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|




























后台回复“生信宝典福利第一波”或点击阅读原文获取教程合集