前文回顾
1. GATK官方教程 / 概述及工作前的布置
2. GATK教程 / 体细胞短变异检测 (SNV+InDel)流程概览
3. GATK教程 / 变异检测前的数据预处理
4. GATK / 体细胞短变异检测工具Mutect2的使用
Mutect2 - 有或没有正常样本配对时,肿瘤体细胞变异检测结果对比Mutect2 - somatic variant calling with/without matched normal sampleHaplotypeCaller is designed to call germline variants, while Mutect2 is designed to call somatic variants.案 例
gatk Mutect2 -R hg38.fa \-I input_tumor.bam -I input_normal.bam \-tumor tumor_sample -normal normal_sample \-pon gatk4_mutect2_4136_pon.vcf.gz \--germline-resource af-only-gnomad.hg38.vcf.gz \--af-of-alleles-not-in-resource 0.0000025 \-L exome_autoXYM.intervals \gatk Mutect2 -R hg38.fa \-I input_tumor.bam \
-pon gatk4_mutect2_4136_pon.vcf.gz \
--germline-resource af-only-gnomad.hg38.vcf.gz \
--af-of-alleles-not-in-resource 0.0000025 \
--genotype-germline-sites \
-L exome_autoXYM.intervals \
-O mt2_unmatched.vcf.gz
后跟 FilterMutectCalls
gatk FilterMutectCalls -R hg38.fa \-V {input.vcf} \
-O {output.fv_vcf}
依赖项
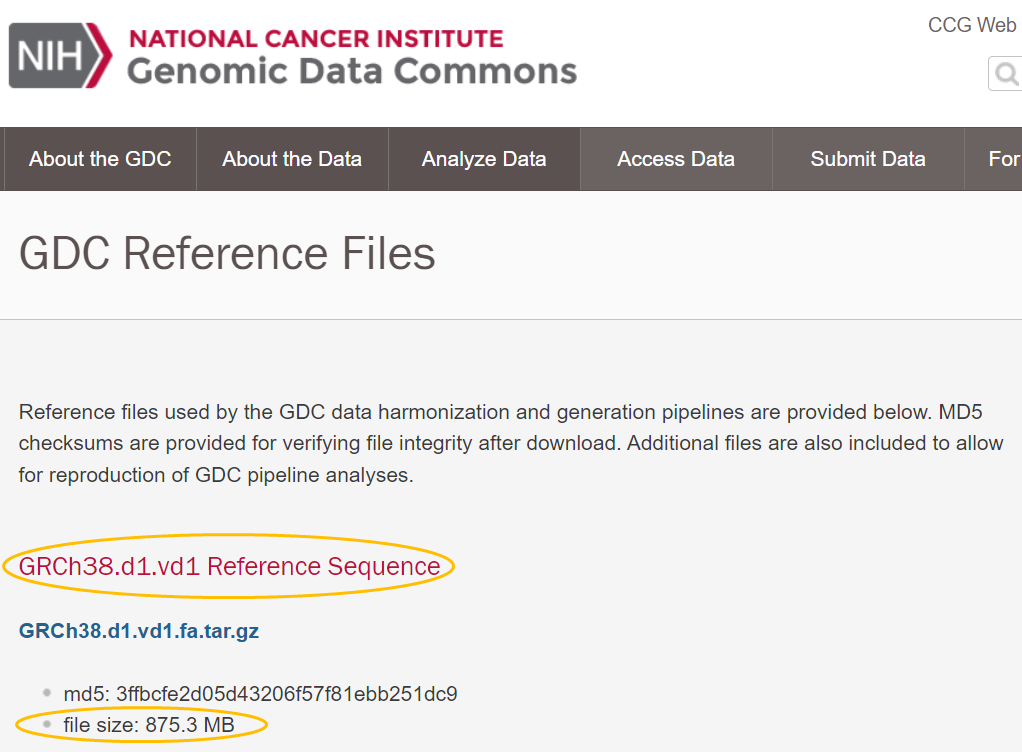
· 间隔/Interval文件使用bedtools(merge)创建,来自RefGene.txt.tar.gz(UCSC) · 使用了“Genomic Data Commons(GDC)”的参考基因组和PoNGDC参考基因组文件
https://gdc.cancer.gov/about-data/gdc-data-processing/gdc-reference-files
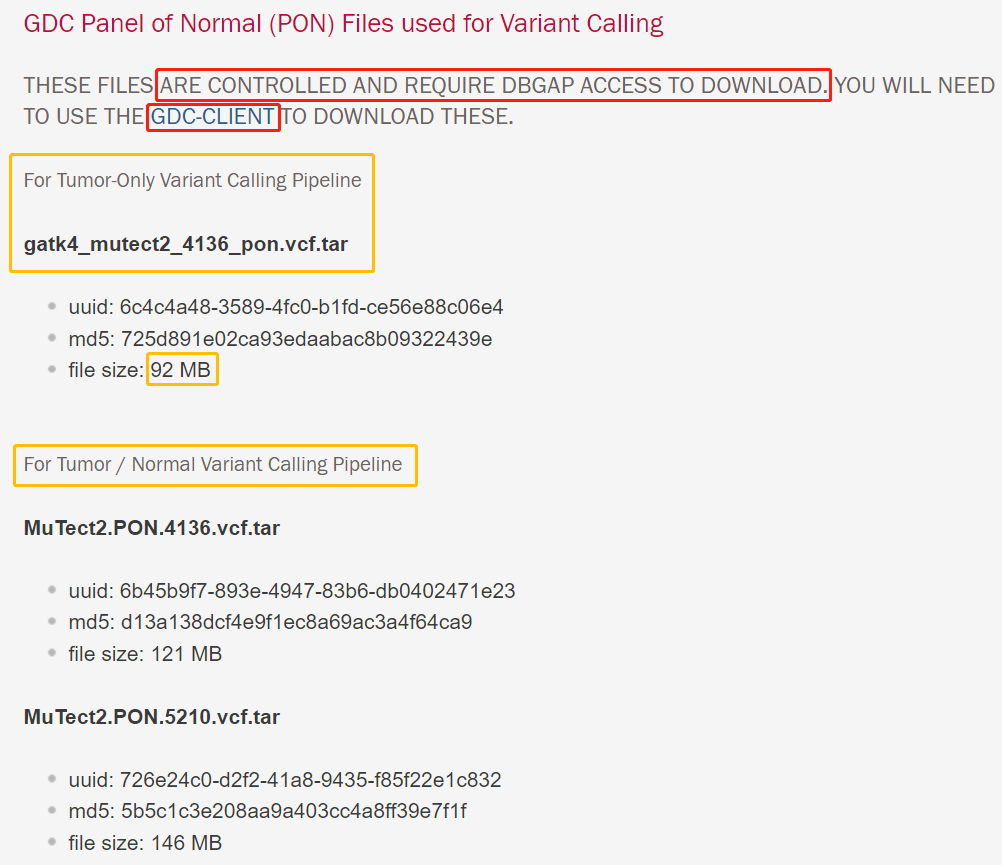
GDC Panel of Normal(PON)文件此PoN文件受控,需要DBGAP访问权限才能下载,且需要使用GDC客户端下载这些内容
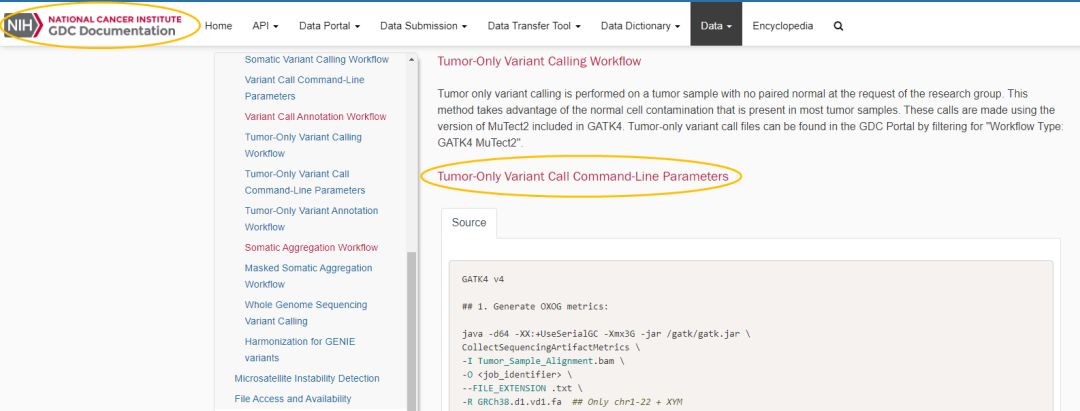
GDC DNA-Seq/Tumor only variant calling workflowhttps://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/DNA_Seq_Variant_Calling_Pipeline/#tumor-only-variant-calling-workflow
问题描述
使用基因组数据共享(Genomic Data Commons, GDC)上可用的测序数据,对Mutect2进行了匹配/不匹配正常样本的测试(Mutect2 with/without matched normal sample)。使用上面的命令,用两种不同的方法检测变异,并进行比较,看看它们重叠的程度如何。 为了做到这一点,在运行FilterMutectCalls后,过滤并获得了具有“PASS”的变异,结果看起来像这样 (对于1个肿瘤患者):
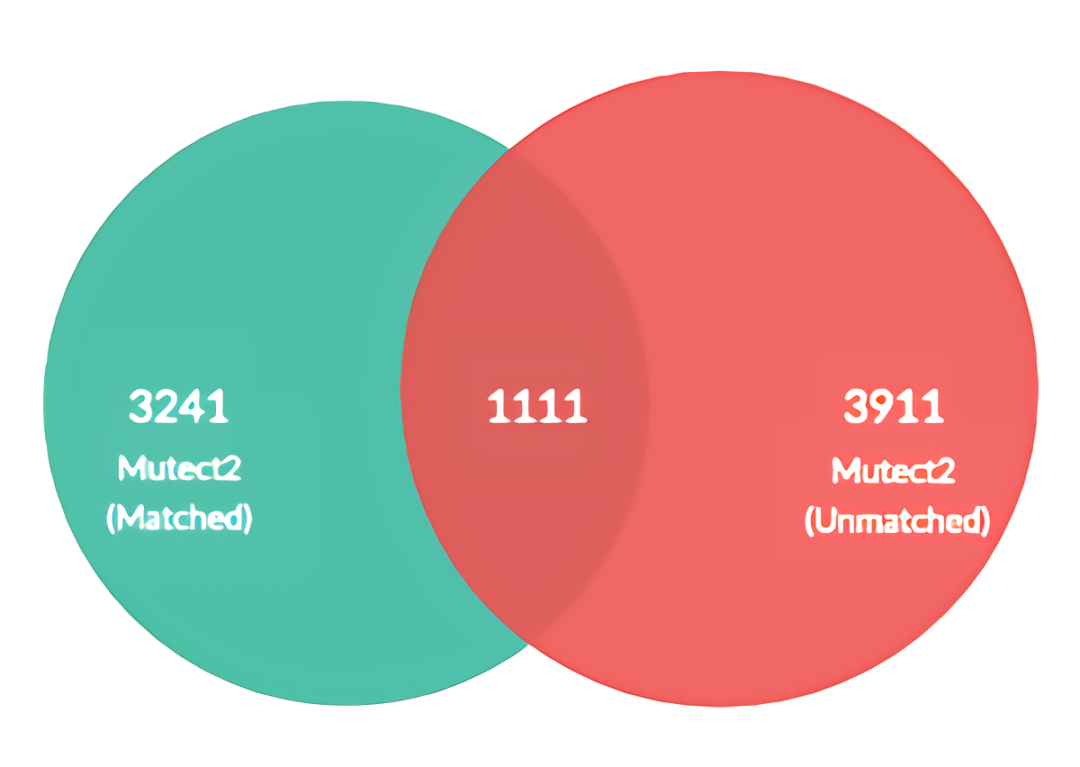
可以看到重叠非常低(每组约20~25%)。这个结果还是很令人担忧的,因为样本均来自同一个肿瘤患者的同一次测序结果。只是:左侧有癌旁(或血液白细胞)测序对照(Matched);右侧只测序了肿瘤组织(Unmatched)。 因为我将使用来自只有肿瘤样本的变异检测流程(Tumor-only variant calling pipeline)的数据。即只对肿瘤样本测序,没有正常样本测序,更没有肿瘤病人自身的正常组织配对样本。每个肿瘤病人都有自身配对的癌旁或血液白细胞测序,以尽可能地获取与肿瘤有关的突变过滤掉病人自身存在的良性的、遗传的、等其它与肿瘤无关的变异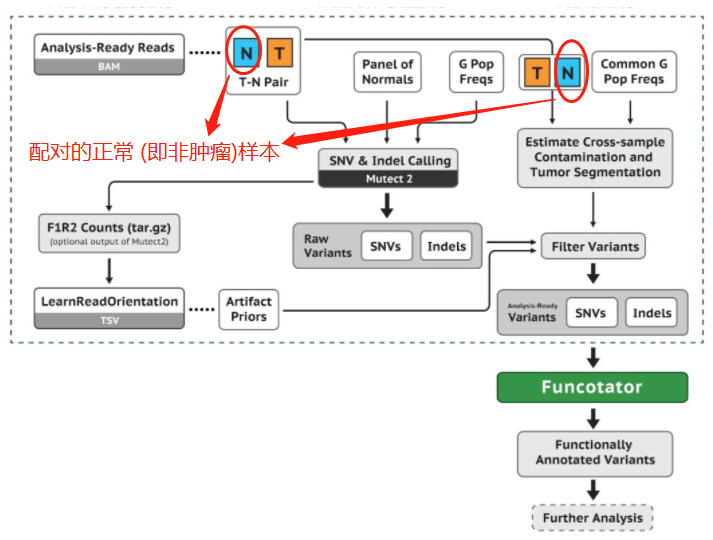
此类问题(无Normal配对的Tumour测序)可能没有明确的解决方案,因为有1个匹配的正常样本(A matched normal sample)才是被官方推荐的方法。 问题是:当一个病人只有肿瘤样本测序可用时(未对该病人的癌旁或血细胞测序),是否有任何额外的过滤(Filtering)技术可以用来减少结果中假阳性(False positives)突变的数量?问题的讨论
GATK支持团队专注于解决工具产生的与GATK工具相关的错误和异常结果的问题。对于所有其它问题,比如这个问题,我们正在构建一个待办事项列表,等我们有能力的时候来解决。请继续发布您的问题,因为我们将挖掘它们以改进文档、资源和工具。我们不能保证得到回复,但是,如果你知道答案,我们会请求其它社区成员帮助。了解详情,请查看我们的支持政策:https://broadinstitute.zendesk.com/hc/en-us/articles/360038469272-What-types-of-questions-will-the-GATK-frontline-team-answer- 你好大卫,抱歉一直拿我的问题打扰你。我之前和GDC的人讨论过,他们用4000+血液正常样本用GATK4 (v4.0.4.0)创建PoN。https://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/DNA_Seq_Variant_Calling_Pipeline/#tumor-only-variant-calling-workflow GDC Panel of Normal (PON) Files used for Variant Calling (gatk4_mutect2_4136_pon.vcf.tar,文件受控制,可能需要dbGAP访问权限才能下载。也需要使用GDC客户端下载),信息来源:https://gdc.cancer.gov/about-data/gdc-data-processing/gdc-reference-files 另外,我只关注乳腺癌,所以我想知道是否应该只纳入PoN上的女性。 如果有人想知道其它样本是否也是如此,我已经尝试了一些其它匹配的数据,并看到了类似的数字(10-15% overlap between tumor-only and matched normal/tumor workflow)。 我还使用HaplotypeCaller确认了非匹配肿瘤分析流程所特有的变异(Variants unique to unmatched tumor-only workflow)不是胚系变异。下面是我用于胚系变异检测(Germline calling)的命令(仅为了测试任何潜在的胚系变异,不做任何过滤):gatk --java-options HaplotypeCaller -R hg38.fa -I normal.bam -O normal.vcf.gz 老实说,你所能希望实现的最好的Tumor-only calling是一组候选变异(A set of candidate variants),其中大部分实际上是胚系变异(即与参考基因组不同的、生来就有的变异,与后天产生的肿瘤体细胞变异无关)。即使你极其保守,不计频率地移除gnomAD中的每1个等位基因(Removed every allele in gnomAD regardless of frequency),仍然会留下数万个独特的胚系变异。 在Low-VAF (Variant allele frequencies)亚克隆和混入大量正常(即非肿瘤细胞)DNA的不纯样本的情况下(这个在临床上几乎无法避免:穿刺活检等方法取出来的组织会存在大量的非肿瘤细胞),你想要的变异的等位基因分数(Allele fraction)与1/2的二倍体杂合(Diploid het)的分数显著不同,FilterMutectCalls可以做得更好(但在胚系变异的检测结果中,也存在着大量偏离50%的分数)。然而,总的来说,Tumor-only calling和Matched normal calling之间的重叠度小,是不可避免的。 罕见的胚系变异,是(有或无Normal配对的)差异的一个来源。它们并不一定会导致Tumor-only calls通过Matched normal来实现过滤。更常见的情况是,罕见的胚系变异的存在,迫使FilterMutectCalls变得保守(Conservative),并过分地过滤了等位基因分数(或比例)接近1/2的真正的体细胞变异(如此高频的体细胞突变可能非常重要)。因为杂合子的胚系/遗传变异的等位基因分数的理论值也是1/2。区别只是在细胞内与细胞间,即:等位基因分数接近1/2的体细胞变异,在一部分细胞中完全是野生纯合子;而胚系/遗传变异的杂合子在所有细胞均为杂合。因此,Bulk DNA-seq无法区分上述两种情况。可能只有单细胞DNA-Seq测序技术可以完成所谓的“Tumor-only calling”(不必采样癌旁或血细胞来Match),这是由体细胞突变的特点所决定的,即一部分细胞正常、一部分细胞突变,单细胞测序恰好可以观察不同细胞中的情况,而胚系突变在身体所有细胞中的情况理论上完全一致。 (有或无Normal配对的)差异的另一个来源,此差异总是导致在Normal(或在HaplotypeCaller的输出中显示的)中不存在的Tumor-only calls,(差异的来源)是可以从Matched normal中检测到的Mapping artifacts。其基本思想是,差异的基因组具有不同的结构变异(SVs)和其它影响(基因组)比对错误(Mapping error)的变异。例如,在着丝粒参考序列间隙(Centromere reference gap)中的一个SNP可能会使“天平”偏向于基因组其它地方的一个比对错误。在某种程度上,导致这一现象的变异是常见的,一个PoN可以并且确实有助于(解决)这类问题,但若有足够多的罕见变异,这就还不够。 我突然想到,尽管我们从未尝试过,但使用父系和母系样本作为两个匹配的正常样本(Mutect2可以通过指定-I表示肿瘤和两个正常样本,以及指定-normal表示两个正常样本来做到这一点)可能会有很大帮助。当然,如果你没有一个Matched normal,也就可能没有来自其父母的正常样本。 这个PoN听起来不错,我觉得没有理由排除男性(研究乳腺癌时)。 你说你在Mutect2 tumor-only calling特有的变异中没有看到胚系突变?我很好奇,你是怎么运行HaplotypeCaller的?以我的理解,用HaplotypeCaller很难捕捉到罕见/独特的胚系事件(我想应该叫单例/Singletons吧?),因为它的后续GenotypeGVCFs使用多个样本中的胚系突变来提升可信度。 David,你说“罕见的种系变异的存在迫使FilterMutectCalls变得保守,并且过度地过滤了等位基因比例接近1/2的真实体细胞变异”。Mutect2如何首先检测到这些罕见的胚系变异,从而知道要过度过滤? 你介意再详细解释一下映比对错误(Mapping error)吗?以及PoN是如何装配和使用的。 Mutect2检测罕见的胚系变异的方式,和检测其它变异的方式一样。关键是,它们非常罕见,甚至在gnomAD中都没有,所以没有任何先验知识(Prior knowledge)表明它们是胚系。(是的,gnomAD并没有完整地收录所有个体的、所有可能会发生的胚系变异;胚系变异完全不断地、随机地发生,3千万个外显子位点的、所有可能的SNP至少有9千万种,而对于InDel的数量更是无法估计) 比对错误(Mapping error)是指:将基因组的一个部位的Reads,对比到了基因组的另一个位置(这可能是由于参考序列不完整、结构变异和同源性造成的)。因为它们是真正的DNA序列,你不能用从测序和样品制备中检测错误的方式,来检测它们。我们可以寻找一些特征(Signatures),但一组正常样本(A panel of normals)也很有帮助,因为(从一个人到另一个人)这些错误往往发生在相同的地方。 我明白了。我以为gnomAD只在FilterMutectCalls中用于胚系过滤器作为先验/Prior?我还以为,如果在人群数据库中没有找到候选变异,就有方法来估计先验? 你也说过,“它们并不一定会导致Tumor-only calls通过Matched normal来实现过滤”。我有点糊涂了,你是说这些罕见的胚系事件不能用匹配的正常样本来过滤掉吗?你是说,一般来说,Mutect2即使有匹配的正常样本,也需要有人群数据库Prior才能过滤掉胚系事件吗? 如果你能详细说明Mutect2 +过滤器/Filters中的哪个统计模型中的哪些参数受到了影响,那将非常有帮助,因为我也在试图理解其中的方法论。 嘿,vctrymao,除了我在评论中提到的HaplotypeCaller命令,其它都没有运行。就像你说的,一般建议(GATK最佳实践)将多个样本一起运行,作为胚系变异检测流程的一部分。然而,为了解决我当时的问题,我决定对少量样本单独运行HaplotypeCaller,不进行任何过滤,以保留所有变异。 对于你的问题关于人群(数据库)资源的使用,对过滤胚系事件,我想指的是下面的链接下部分的“A variant allele in the case sample is not called if the site is variant in controls”/“如果某个位点在对照/Controls中变异,将不会从疾病/Case样本中调用/Call该变异的等位基因”(在底部)将有帮助:https://gatk.broadinstitute.org/hc/en-us/articles/360035890491-Somatic-calling-is-NOT-simply-a-difference-between-two-callsets#:~:text=HaplotypeCaller%20is%20designed%20to%20call,designed%20to%20call%20somatic%20variants. 如果你决定进行任何与这篇文章相关的测试,请务必更新! vctrymao,你是对的,来自胚系(变异数据库)资源的等位基因频率被用作Prior。 如果一个变异不在胚系(变异数据库)资源/Germline resource中,我们会指定一个比“1/(胚系资源的大小)”更罕见的默认等位基因频率(Allele frequency)。也就是说,如果你的10万个二倍体样本的种系资源中没有某个等位基因,我们可以猜测频率小于“1/20万”。 我所说的“它们并不一定会导致Tumor-only calls通过Matched normal来实现过滤”,我的意思是,即使在Tumor-only模式下,罕见的胚系变异有时也会被过滤掉。 Mutect2应该总是与胚系变异资源一起运行,即使是在Matched normal模式下,尽管它被设计为在没有胚系资源的情况下尽可能地运行良好。 1. 即使是在tumor-only模式下,如果罕见的胚系变异有时也会被过滤掉,这是好事,不是吗?所以问题是,罕见的胚系变异仍然没有被过滤掉?那些确实被过滤掉的胚系突变的特征是什么? 2. 你是说大多数胚系突变会在gnomAD这样的库中被捕获,所以成为大量候选胚系突变的Prior? 3. 你还说“罕见胚系变异的存在迫使FilterMutectCalls变得保守,并且过度过滤了等位基因比例接近1/2的真正的体细胞变异。”我到现在还不明白这是怎么回事。稀有胚系变异的哪一方面迫使FilterMutectCalls变得保守?在我看来,罕见的胚系变异和常见的胚系变异之间唯一的区别是人群频率Prior。但既然体细胞突变也没有人群Prior,你是说正因为如此,Mutect2把所有VAF为1/2的都称为胚系吗? 4. 在matched-normal calls而非tumor-only calls中存在哪些Calls?在什么情况下,matched-normal能够帮助识别出tumor-only caller看不到的体细胞变异? 5. HaplotypeCaller能发现罕见的胚系变异吗?我想知道你是否可以抵消我在“3)”中的观点,通过允许VAF接近1/2的体细胞变异通过过滤器,然后通过HaplotypeCaller过滤掉所有剩余的(和罕见的)胚系变异。 嘿,vctrymao,关于#4,David Benjamin在之前的评论之一中涵盖了潜在的情形:https://gatk.broadinstitute.org/hc/en-us/community/posts/360057810051/comments/360009638892 1. 即使是在tumor-only模式下,如果罕见的胚系变异有时也会被过滤掉,这是好事,不是吗?所以问题是,罕见的胚系变异仍然没有被过滤掉? 这完全取决于等位基因分数与①体细胞聚类模型确定的谱/Spectrum的匹配程度,与(Versus)②局部拷贝数给出的胚系等位基因频率的匹配程度(如果使用CalculateCondition的-tumor-segmentation输入;否则,假设每个地方的拷贝数都是2)。 2. 你是说大多数胚系突变会在gnomAD这样的库中被捕获,所以成为大量候选胚系突变的Prior? 是的,但问题是,这种罕见的胚系变异占了胚系变异的很大一部分。相反,罕见的胚系变异比体细胞变异更常见。 3. 你还说“罕见胚系变异的存在迫使FilterMutectCalls变得保守,并且过度过滤了等位基因比例接近1/2的真正的体细胞变异。”我到现在还不明白这是怎么回事。稀有胚系变异的哪一方面迫使FilterMutectCalls变得保守? 在我看来,罕见的胚系变异和常见的胚系变异之间唯一的区别是人群频率Prior。但既然体细胞突变也没有人群Prior,你是说正因为如此,Mutect2把所有VAF为1/2的都称为胚系吗?4. 在matched-normal calls而非tumor-only calls中存在哪些Calls?在什么情况下,matched-normal能够帮助识别出tumor-only caller看不到的体细胞变异? 1个匹配的正常样本(Matched normal)可以提供非常好的证据,证明1个变异肯定不是1个胚系变体。(A matched normal can give very good evidence that a variant is definitely not a germline variant.)5. HaplotypeCaller能发现罕见的胚系变异吗? 我想知道你是否可以抵消我在“3)”中的观点,通过允许VAF接近1/2的体细胞变异通过过滤器,然后通过HaplotypeCaller过滤掉所有剩余的(和罕见的)胚系变异。
你可以这样做,但我看不出这样做会有什么效果。HaplotypeCaller无法区分具有大的等位基因分数(Large allele fractions)的体细胞变异与胚系变异。 你好,我在哪里可以下载到这个GATK中的文件, gatk4_mutect2_4136_pon.vcf.gz,能给我一个链接吗?谢谢资料来源
https://gatk.broadinstitute.org/hc/en-us/community/posts/360057810051-Mutect2-somatic-variant-calling-with-without-matched-normal-samplehttps://gatk.broadinstitute.org/hc/en-us/community/topics/360001488872-Somatic往期精品(点击图片直达文字对应教程)
机器学习
后台回复“生信宝典福利第一波”或点击阅读原文获取教程合集